
Bioinformatics Services
We provide training and service opportunities across a variety of bioinformatics techniques. We work with genomes and transcriptomes, including, but not limited to de novo assemblies, resequencing, differential gene expression analyses, differential peak calling, variant discovery, and annotation. Custom services available on a case-by-case basis.
Whole genomes
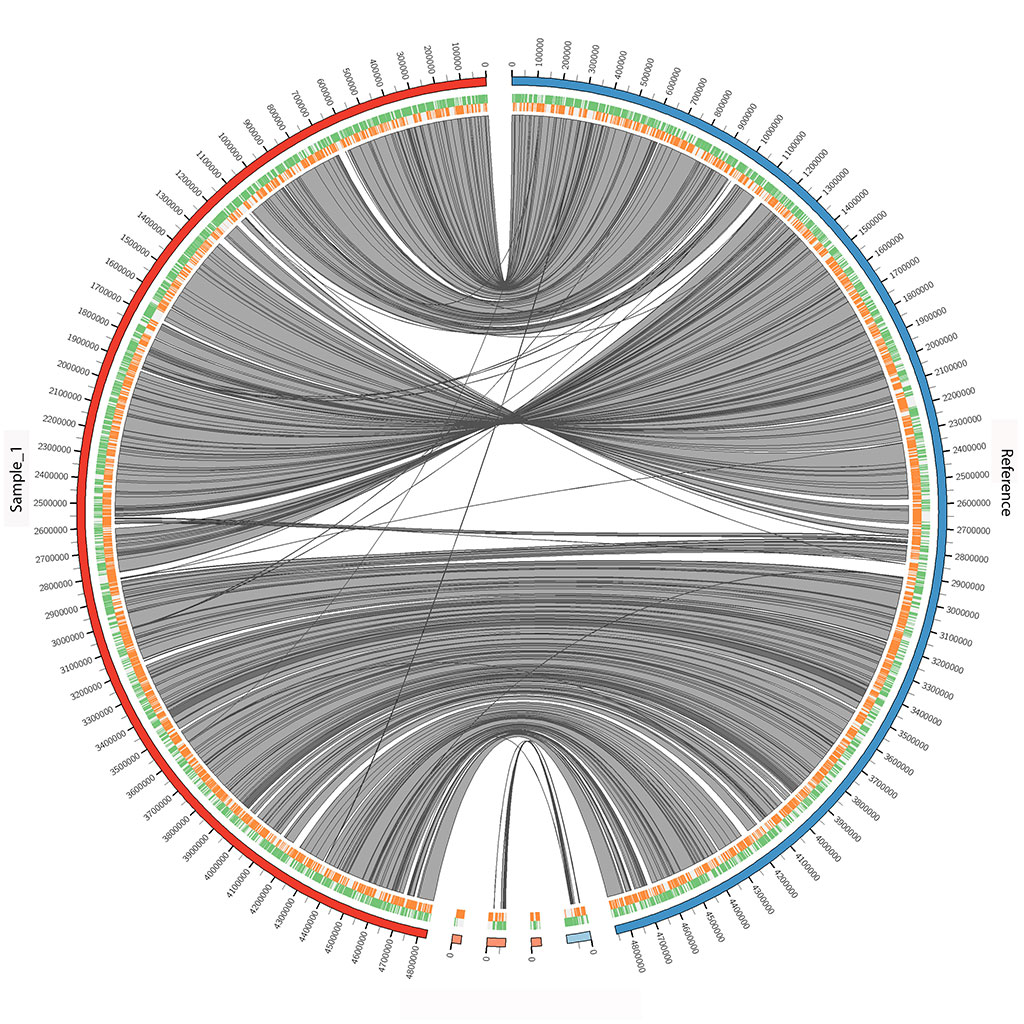
Experienced in de novo assemblies and resequencing, using next-generation sequencing long and/or short read technologies. We move datasets from raw reads through assemblies/alignments, gene and functional annotation, pathway analysis, variant discovery (single nucleotide polymorphisms, insertions/deletions, breakpoints, rearrangements, inversions, and copy-number variation), and selective sweeps.
We also have experience in comparative genomics using various techniques, such as genome-wide association studies and comparisons of other variant discovery methods mentioned above.
Finally, we have used methods to examine the subset of the genome known as the exome including single samples, family trios, and matched tumor/normal pairs
Transcriptomics “common analyses”
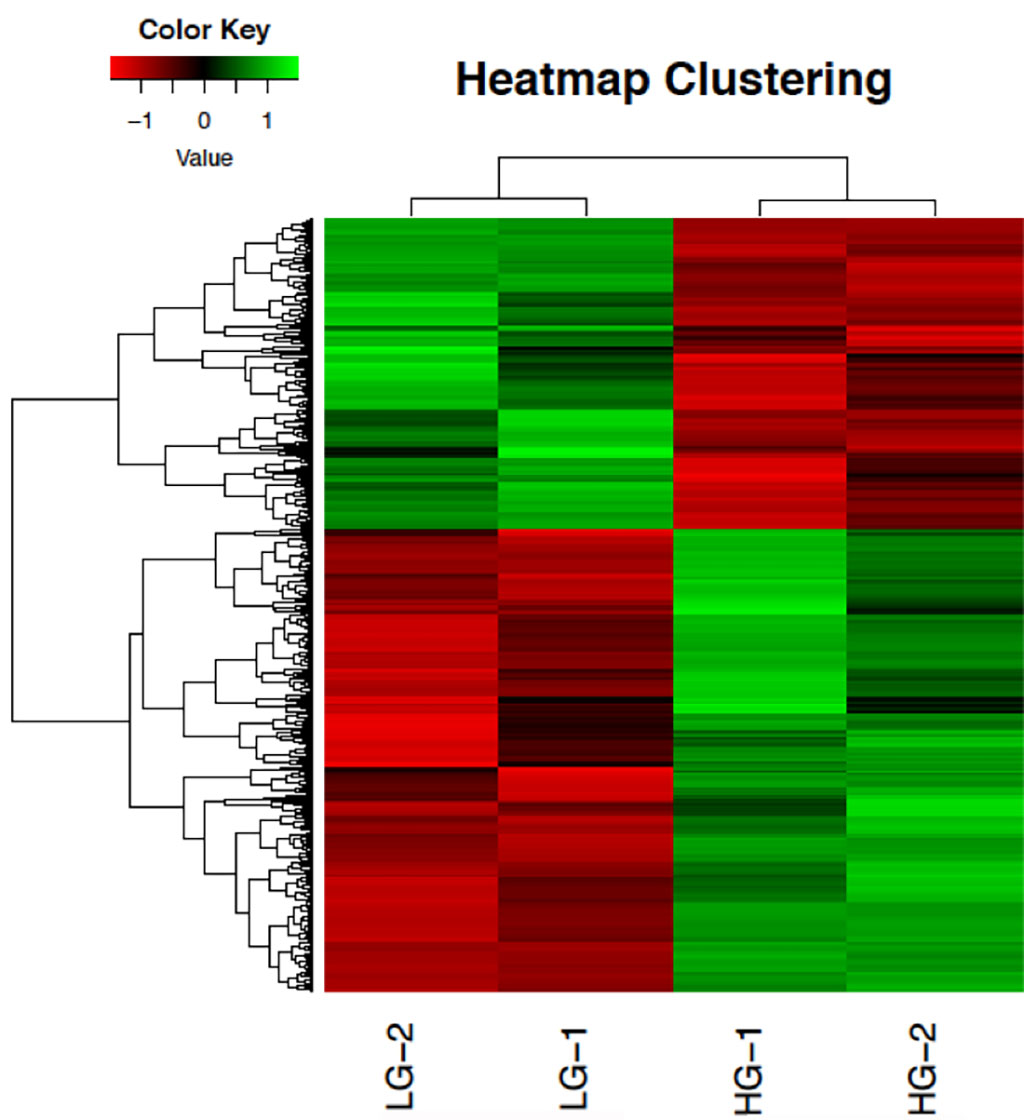
We have completed projects with RNAseq, including small RNA profiling, differential gene expression (DGE) studies using de novo assembled transcriptomes and available reference sequences, and gene co-expression network construction. Gene identification and functional annotation are done for de novo assemblies. Results from DGE studies include gene ontology terms, pathway enrichment and information from the KEGG pathway database.
Microbiomics
We perform the taxonomic and functional profiling of both host associated and environmental microbial communities based on amplicon sequencing and multi-omics approaches.
Our barcoding methods (16S and ITS) describe the composition of the community structure, its alpha and beta diversity, and associated multivariate analysis.
Additionally, we can profile the taxonomic composition and metabolic potential of microbiomes through genome-resolved metagenomics, metagenomics, and metatranscriptomics. By combining these approaches and applying well established and customized databases and pipelines, we can describe the communities at different levels. Examples: read and ORF/CDS/transcript abundances, recovery of Metagenome Assembled Genomes (MAGs), differential expression, and more.
Palmetto Resources
CUGBF currently owns one high memory node and a 75 terabyte (TB) file system on Clemson University’s Palmetto cluster, which is supported by the research computing and data (RCD) team. Our node is equipped with 80 cores and 1.5 TB of random-access memory (RAM).
Our facility maintains hundreds of software programs for most types of bioinformatics analyses. A list grouped by categories can be found here, and a full list (with version numbers) is available here.
The software we maintain is accessible through modules on the Palmetto cluster. In order to use these programs, you must have a Palmetto account. If you need to apply for one (free to all Clemson faculty, staff, and students), please follow this link.
RCD also provides introductory and advanced training throughout the year, and their workshops are available here.
A great resource with additional information on the Palmetto cluster is their online user guide.
Once you have a Palmetto account, please agree to and sign our acknowledgement agreement in order to be given access to our software modules.
Contact us for a copy of our acknowledgment agreement.
“We had an excellent experience with the facility and service. We will come back to you in the future.”
— Dr. Venugopal Mendu
Department of Plant and Soil Science
Texas Tech University