Research

Charge Separation and Electron Injection Dynamics
The efficiency of electron injection into the conduction band in these materials is strongly influenced by charge separation and recombination dynamics, which are modulated by ligand nuclear motion and interfacial structure. This research direction focuses on electronic structure and molecular dynamics methodologies capable of describing ligand-dependent charge transfer pathways. By connecting nuclear motion with nonequilibrium electronic behavior, this work aims to provide a mechanistic understanding of how ligand environments control photovoltaic performance. These insights guide the rational design of next-generation solar energy materials with improved efficiency, stability, and tunability.
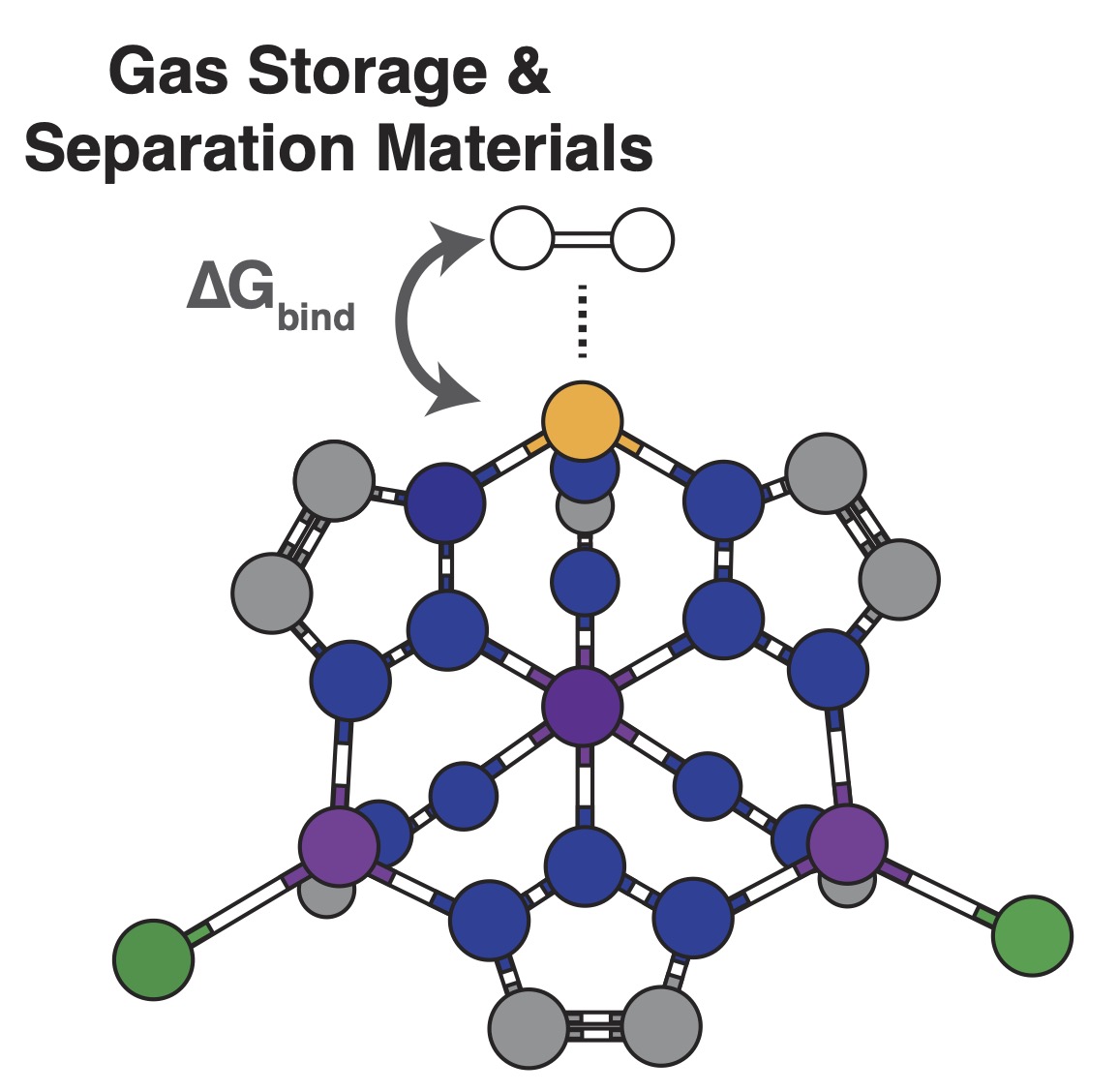
Improving Thermodynamic Predictions of Adsorbate Binding
Nanoporous frameworks are promising, cost-effective materials for small-molecule gas separation and storage, offering a potential sustainable alternative to fossil-fuel–based processes. Despite this promise, reliably predicting the thermodynamics and transport of adsorbed species within these materials remains a major challenge for current computational approaches, particularly for light and highly mobile species such as water and isotopologues like tritiated water. This research direction focuses on developing vibrationally resolved theoretical methods to accurately describe adsorbate binding thermodynamics and transport in confined environments. These advances aim to enable more predictive modeling of adsorption and diffusion processes, supporting the design of nanoporous materials for efficient room-temperature gas separation, storage, and isotope transport applications.

Excited-State Electronic Structure and Nonadiabatic Dynamics
Making reliable predictions of coupled electronic and nuclear dynamics remains challenging due to the cost and limitations of current electronic structure and molecular dynamics methods. A central difficulty in quantum dynamics is the so-called “curse of dimensionality,” which arises from the strong coupling between electronic and nuclear degrees of freedom in vibronic systems. This research direction develops improved electronic structure methods, including orbital-optimized time-dependent density functional theory (TDDFT), coupled with quasi-classical approaches to molecular dynamics such as SQC/MM. The goal is to improve the description of electronic structure in excited states while providing more efficient and physically transparent treatments of electronic–nuclear coupling. These advances aim to enable more accurate simulations of nonadiabatic dynamics and improved interpretation of vibronic spectra and ultrafast molecular processes.