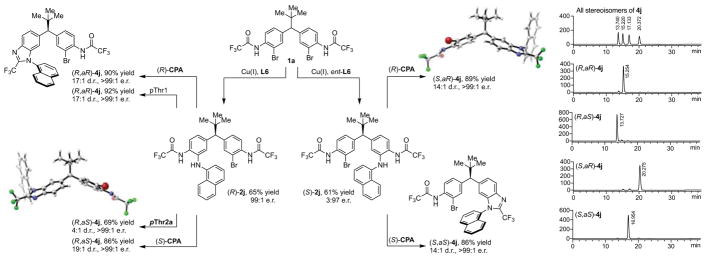
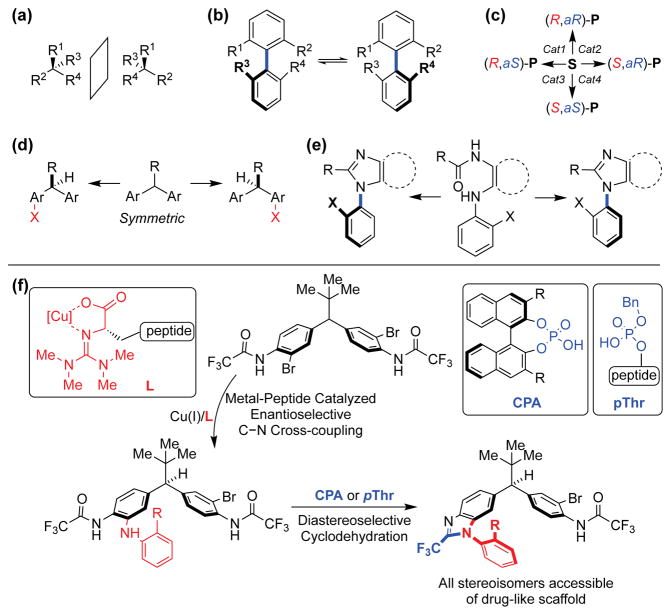
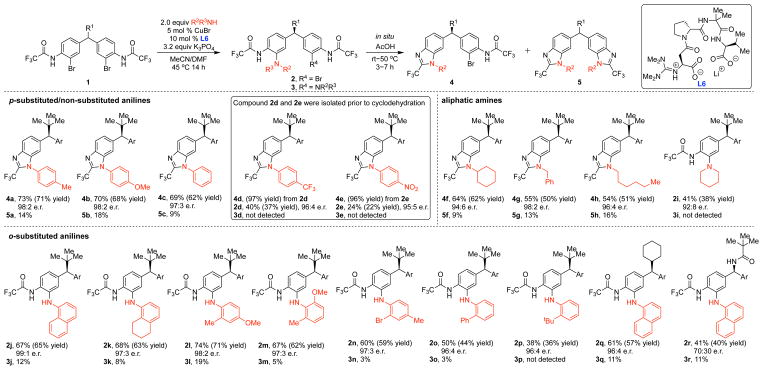
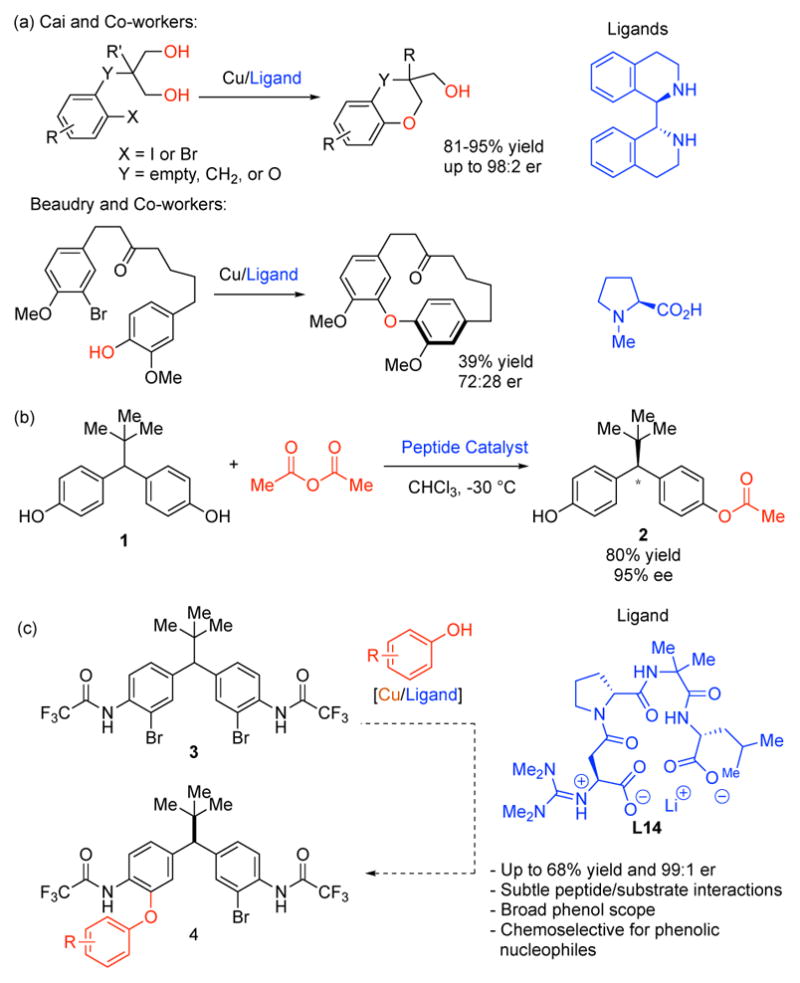
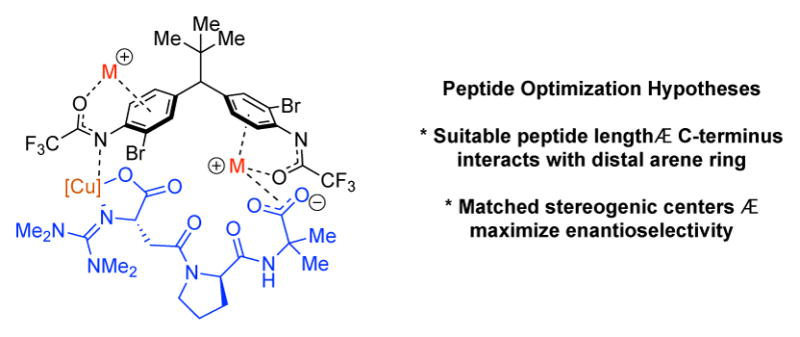
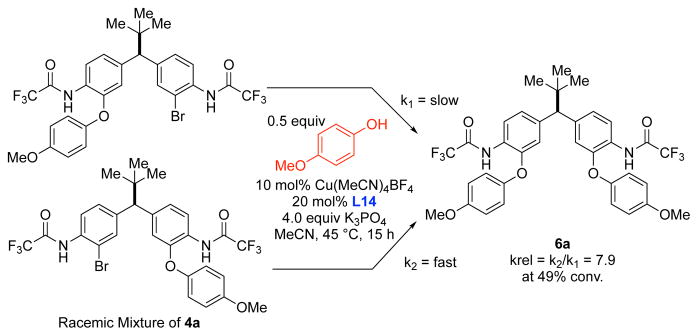
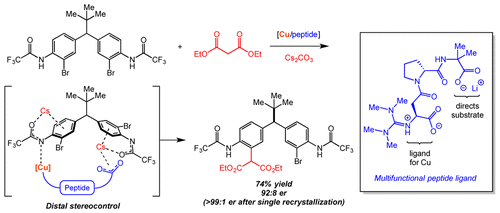
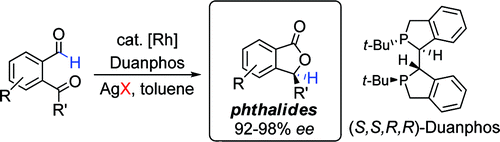
A Stereodynamic Redox-Interconversion Network of Vicinal Tertiary and Quaternary Carbon Stereocenters in Hydroquinone-Quinone Hybrid Dihydrobenzofurans
9. A Stereodynamic Redox-Interconversion Network of Vicinal Tertiary and Quaternary Carbon Stereocenters in Hydroquinone-Quinone Hybrid Dihydrobenzofurans Storch, G.; Kim, B.; Mercado, B. Q.; Miller, S. J. Angrew. Chem. Int. Ed. 2018, 57, 15107-15111.
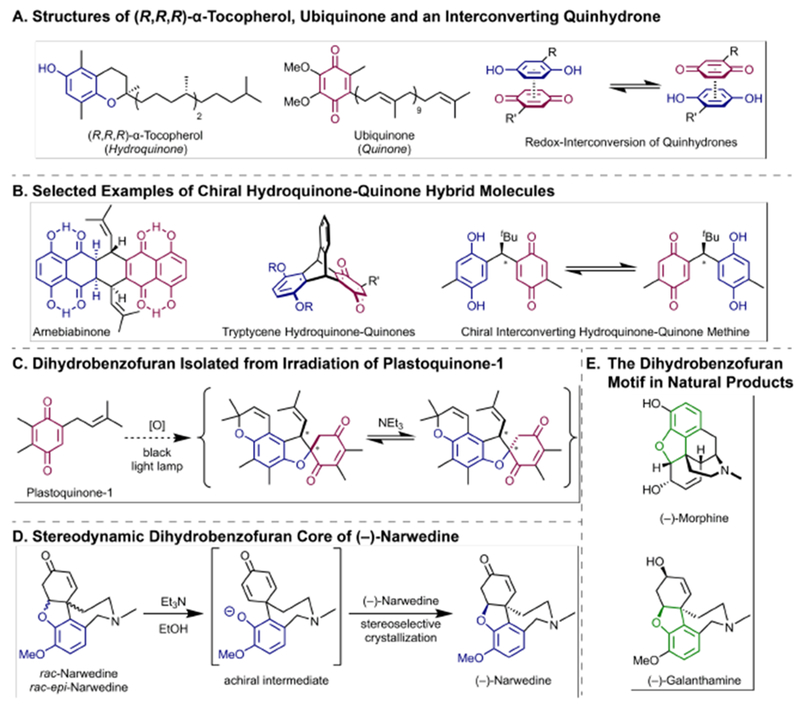
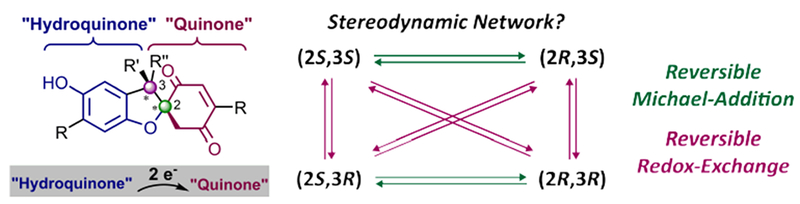
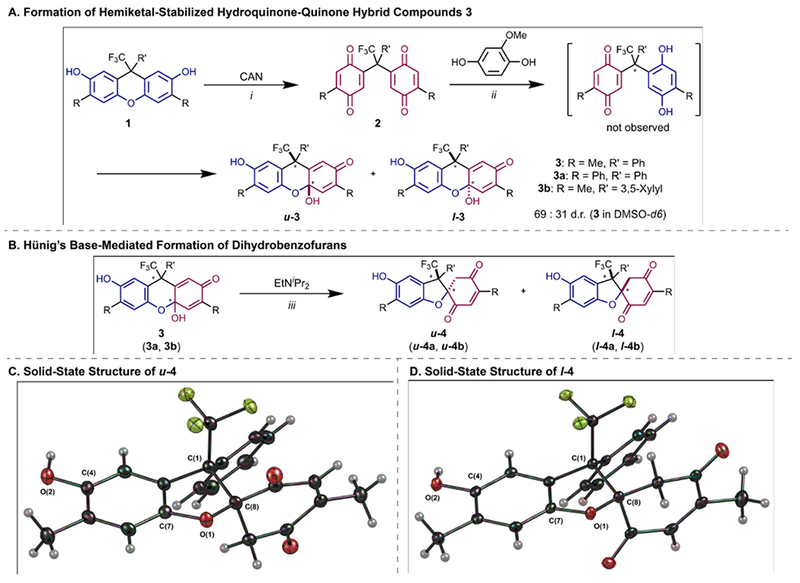
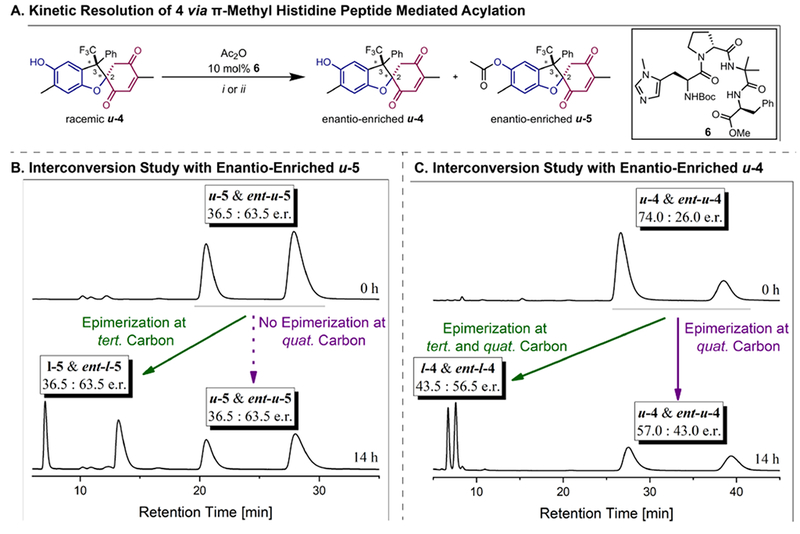
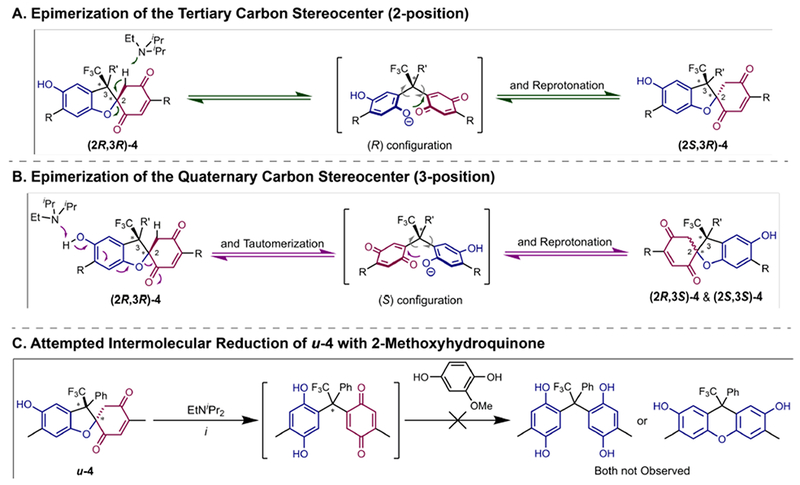
Reversible redox processes involving hydroquinones and quinones are ubiquitous in biological reaction networks, materials science, and catalysis. While extensively studied in intermolecular settings, less is known about intramolecular scenarios. Herein, we report hydroquinone-quinone hybrid molecules that form two-stereocenter dihydrobenzofurans via intramolecular cyclization under thermodynamic control. A π-methylhistidine peptide-catalyzed kinetic resolution allowed us to study the stereodynamic behavior of enantio- and diastereo-enriched dihydrofurans. In the course of this study, it was revealed that a reversible intramolecular redox-interconversion network connects all four possible stereoisomers via inversion of a quaternary carbon stereocenter without achiral intermediates. As a result, these findings on hydroquinone-quinone hybrid molecules provide insights into potential natural origin and synthetic access of the common dihydrobenzofuran motif.
Public Access »
View on PubMed »